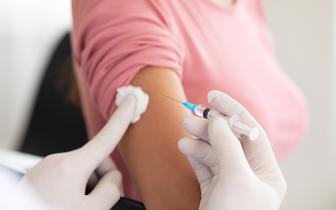
Co trzeba wiedzieć o biopodobnych produktach leczniczych
Koalicja organizacji pozarządowych zainteresowanych zagadnieniem leków biologicznych i biopodobnych, zorganizowała w warszawskiej siedzibie Komisji Europejskiej debatę na temat preparatów biopodonych. Wśród uczestników znaleźli się m.in. przedstawiciele polskiego rządu, Komisji Europejskiej, stowarzyszeń firm farmaceutycznych, stowarzyszeń pacjentów oraz eksperci w zakresie prawa i medycyny.
Podstawą do dyskusji był opracowany w 2013 roku dokument informacyjny — konsensus Unii Europejskiej „Co należy wiedzieć o biopodobnych produktach leczniczych?”. Rozpoczynając dyskusję, Thomas Heynisch z Dyrekcji Generalnej Komisji Europejskiej ds. Przedsiębiorstw i Przemysłu poinformował, że najważniejszym celem dokumentu było wyjaśnienie, czym są leki biopodobne oraz jakie warunki muszą spełniać, aby zostały zarejestrowane i dopuszczone do obrotu. Ponieważ zarządzanie i podejmowanie decyzji w zakresie ochrony zdrowia należy do kompetencji rządów poszczególnych krajów Unii, rozpowszechnienie leków biopodobnych w krajach członkowskich jest mocno zróżnicowane. Różny jest też poziom wiedzy na temat tych leków nawet wśród osób wykonujących zawody medyczne.Procedura rejestracyjna i nazewnictwo Zdaniem prof. Piotra Fiedora, reprezentującego Europejską Agencję Leków (EMA), dostęp lekarzy do wiedzy i informacji o lekach biologicznych jest bardzo ważny. Według obowiązujących procedur, proces badań klinicznych i rejestracji leków biopodobnych trwa zwykle rok lub dwa lata. Wyniki tych badań są prezentowane Komisji Europejskiej w celu podjęcia decyzji o wprowadzeniu leku do obrotu. Opinia zawarta jest w Charakterystyce Produktu Leczniczego (ChPL), czyli dokumencie, który mówi o wskazaniach klinicznych, przeciwwskazaniach, działaniach niepożądanych, możliwości stosowania leku poza wskazaniami na odpowiedzialność lekarza. Leki biopodobne są rejestrowany wyłącznie w procesie rejestracji centralnej przez Europejską Agencję Leków. Reprezentujący Stały Komitet Lekarzy Europejskich (CPME) dr Bernard Maillet wskazał na trzy najważniejsze kwestie związane z lekami biopodobnymi, tj. bezpieczeństwo i skuteczność kliniczną, zastępowalność oraz dostęp lekarzy do informacji. W jego opinii, ważne jest nazewnictwo leków biopodobnych, aby nazwa międzynarodowa była inna niż leku referencyjnego. Wskazał też, że wiedza o lekach biologicznych i biopodobnych przekazywana przyszłym lekarzom przez uczelnie medyczne jest dalece niewystarczająca.Irena Rej, prezes Izby Gospodarczej „Farmacja Polska”, poinformowała, że Izba od dawna zabiega o sformułowanie definicji leków biologicznych w polskim systemie prawnym. Definicja taka nie została wprowadzona, według Ireny Rej, z powodów finansowych, a jej brak ma negatywne implikacje — utrudnia m.in. uporządkowanie metod prowadzenia pharmacovigilance.Maciej Wieczorek w imieniu Polskiego Związku Pracodawców Przemysłu Farmaceutycznego podkreślił, że proces rejestracji leków biologicznych zapewnia skuteczność i bezpieczeństwo ich stosowania. „Często badania kliniczne leków biopodobnych są tak zaprojektowane, że dają więcej informacji na temat ich bezpieczeństwa niż badania rejestracyjne leków referencyjnych, co wynika z ich skali. Statystycznie muszą one być duże” — zaznaczył Maciej Wieczorek. Zwrócił też uwagę, że już dzisiaj leki biologiczne stanowią 40 proc. nowych leków rejestrowanych, a w ciągu najbliższych kilkunastu lat przekroczą połowę nowych rejestracji. Z tego powodu rozwój branży leków biopodobnych jest ważny dla gospodarki. Miejsce w systemie refundacyjnymW opinii Pawła Sztwiertni, dyrektora generalnego Związku Innowacyjnych Firm Farmaceutycznych INFARMA, polski system prawny nie nadąża za rozwojem farmakoterapii i biotechnologii, a w kontekście dostępu do leków biopodobnych jest niespójny. Jako najważniejszą przyczynę tego stanu rzeczy wskazał brak polityki lekowej państwa, jako dokumentu, który powinien wskazać miejsce leków biologicznych i biotechnologii w procesie leczenia. Jego zdaniem, koszty inwestowania w najnowsze technologie, opłacalne z punktu widzenia długoterminowej polityki państwa, są oceniane jako koszty krótkoterminowe i niepotrzebnie ograniczane. Rząd powinien także zdefiniować politykę przemysłową, czyli określić rolę i miejsce przemysłu farmaceutycznego i biofarmaceutycznego, zarówno producentów lokalnych, jak i międzynarodowych firm działających w Polsce. Dyr. Sztwiertnia podkreślił także ogromne znaczenie edukacji lekarzy i pacjentów na temat leków biologicznych, a zwłaszcza rzetelnej informacji dla lekarzy w kontekście autonomii przy podejmowaniu decyzji terapeutycznych, które nie powinny być ograniczane przez rozwiązania systemu refundacji.Wiceminister Igor Radziewicz--Winnicki zaprezentował stanowisko Ministerstwa Zdrowia: „Uznajemy, że dla systemu refundacyjnego jest to ta sama technologia, która pomimo różnic wynikających z rejestracji tych produktów, jest traktowana jako technologia generyczna w odniesieniu do leków referencyjnych”. W kwestii zastrzeżeń środowiska lekarskiego dotyczących ograniczenia autonomii lekarskiej preskrypcji Igor Radziewicz-Winnicki uważa, że uchwalając ustawę refundacyjną Sejm podjął odważną decyzję przyznając wyższość interesu dobra publicznego nad autonomią preskrypcji lekarskiej. „Z dzisiejszej perspektywy jestem przekonany, że była ona słuszna” — dodaje wiceminister.Zdaniem Igora Radziewicza-Win-nickiego, to właśnie harmonizacja wskazań medycznych i podejmowanie decyzji terapeutycznych na poziomie ministerstwa i urzędu rejestracji jest mechanizmem, który pozwala wynegocjować dobre warunki cenowe dla refundacji leków. Jednocześnie powoduje to, że „określone produkty lecznicze są refundowane w zastosowaniach, które wynikają z najlepiej poznanych dowodów naukowych ich skuteczności w określonym stadium rozwoju danej choroby”. Ten mechanizm, choć jest postrzegany przez środowisko lekarskie jako jeden z największych mankamentów ustawy refundacyjnej, generuje jednak ogromny potencjał rozwoju systemu refundacji i pozwala obejmować nim coraz to nowe leki, w tym innowacyjne. Argument o ograniczaniu autonomii preksrypcyjnej lekarzy wiceminister uznaje za ważny, „ale mniej ważny niż argument dobra pacjentów, którzy jednak in gremio zyskują dzięki szerszemu dostępowi do potrzebnych technologii medycznych, które są zastosowane zgodnie z aktualną wiedzą medyczną i zgodnie z najlepiej pojętą zasadą racjonalności prowadzonej farmakoterapii”. Wiceminister dodał, że nie widzi potrzeby odrębnego definiowania pojęcia leku biologicznego w polskim prawie. Granice zamiennościW opinii Ireny Rej z Izby „Farmacja Polska” analiza zamiennictwa leków biologicznych i biopodobnych na przykładzie 7 krajów europejskich pokazuje, że tylko w jednym kraju — Czechach — wpisano rozwiązanie automatycznej obniżki ceny o 15 proc. przy wpisywaniu leku biodopodobnego na listę refundacji. „Praktyka europejska sprowadza się do jednego — wszyscy twierdzą, że automatyczne zamiennictwo jest praktycznie niemożliwe, powiedziałabym nieetyczne” — dodała Irena Rej.Michał Czarnogórski z Urzędu Rejestracji Produktów Leczniczych ocenił, że procedura rejestracyjna, obejmująca szeroki zakres badań, zapewnia bezpieczeństwo stosowania leków biologicznych i biopodobnych. Podstawą do rejestracji każdego leku biopodobnego jest nie tylko badanie biorównoważności, ale także szereg badań porównawczych, które służą wykazaniu różnorakiej równoważności w stosunku do leku referencyjnego, począwszy od badań klinicznych immunogenności, poprzez tolerancję miejscową, interakcje z receptorami aż do dużych badań porównawczych. Według Michała Czarnogórskiego, europejskie władze rejestracyjne nie stwierdziły dotychczas jednoznacznie, że leki biopodobne mogą być stosowane „wymiennie, zmiennie i przestawnie”, pozostawiając tę decyzję władzom narodowym. W jego ocenie, władze narodowe nie mają jednak powodów, aby sprzeciwiać się takiemu stosowaniu leków biopodobnych. Odnosząc się do opinii przedstawicieli Ministerstwa Zdrowia i Urzędu Rejestracji, prof. Piotr Fiedor podkreślił, że nie tylko sama cząsteczka leku ma znaczenie, ale także mechanizm jej działania. W kwestii zamieniania leków zaznaczył, że według wytycznych EMA, o ewentualnej zamianie leku na inny, czyli wymiennik, w trakcie terapii „decyduje przede wszystkim dyskusja pacjenta i lekarza”. O uzasadnionej zamianie terapii można mówić, jego zdaniem, tylko wtedy, gdy lekarz ma do czynienia z uczuleniem albo nietolerancją na produkt leczniczy lub brakiem skuteczności. Lekarz jest jedyną osobą, która może określić korzyść w stosunku do ryzyka stosowania konkretnego leku u pacjenta. Jednocześnie wskazał na praktykę stosowaną w wielu krajach, w których lekarz ma prawo do wyboru droższej terapii, ale musi swój wybór uzasadnić, w przeciwnym razie pacjent traci prawo do refundacji. Ekstrapolacja działań niepożądanych leków jest przedmiotem troski wielu krajów europejskich. Tworzone są duże rejestry pacjentów i jednostek chorobowych, gdzie wymienianie leku biologicznego i leku biopodobnego może mieć znaczenie w ekstrapolacji działań niepożądanych i skutków terapii. EMA dodatkowo stworzyła sieć współpracy pomiędzy agencjami krajowymi w celu wymiany doświadczeń w odniesieniu do działań niepożądanych leków biologicznych. Według prof. Fiedora, taki rejestr powinien być prowadzony także w Polsce, a dane powinny być uwzględniane w ocenie, czy można bezpiecznie wprowadzić zamiennik w terapii lekiem biologicznym. Dorota Karkowska z Instytutu Praw Pacjenta i i Edukacji Zdrowotnej oceniła, że proces implementacji prawa unijnego do polskiego systemu jest niekompletny. W jej opinii, niezbędna jest dobra nowelizacja prawa farmaceutycznego, uporządkowanie definicji oraz szczególny priorytet dla monitorowania bezpieczeństwa farmakoterapii. Lekarze i farmaceuci często zaniedbują obowiązku udzielania informacji pacjentom. Tymczasem według zasad unijnych pacjent musi być poinformowany o zamianie leku biologicznego na inny. Skrytykowała zaburzenie autonomii lekarzy, do którego dochodzi przy preksrypcji aptecznej oraz przy przetargach w szpitalach. Mec. Paulina Kieszkowska-Knapik zauważyła, że w polskim systemie refundacji obowiązuje odwrotna zasada obowiązkowej zamiany leku, wymuszonej przez zmiany limitu refundacji. Potwierdziła też brak monitorowania farmakoterapii, np. w formie rejestrów, wskazanego zwłaszcza w sytuacji przestawiania pacjentów na inny lek, co wobec częstych zmian listy refundacyjnej może wielokrotnie zachodzić w odniesieniu do jednego pacjenta. System refundacji powinien, zdaniem mec. Kieszkowskiej-Knapik przewidywać także przestrzeń dla tych pacjentów, u których zamiana leku nie przyniosła pozytywnych efektów. W ocenie wiceministra Radziewicza-Winnickiego, ważne jest budowanie poczucia bezpieczeństwa stosowania leków biologicznych i biopodobnych na podstawie gruntownej wiedzy naukowej. Nie zgodził się jednak z koncepcją krajowego rejestru stworzonego specjalnie w celu gromadzenia danych dotyczących farmakoterapii lekami biologicznymi. Ważny wskaźnik efektywności kosztowejPrezes Polskiej Federacji Szpitali Jarosław J. Fedorowski zwrócił uwagę na ekonomiczny aspekt wprowadzania leków biopodobnych, zwłaszcza w kontekście warunków finansowych polskich szpitali. Leki biopodobne mogą pozytywnie wpływać na efektywność kosztową leczenia polskich pacjentów, zwłaszcza że nasze szpitale dysponują budżetami co najmniej 8-10-krotnie niższymi niż szpitale w Europie Zachodniej. W związku z tym ich środki przeznaczane na farmakoterapię są również niższe, co musi wpływać na udzielane świadczenia, standard opieki i możliwości działania personelu medycznego. W ocenie Jarosława Fedorowskiego, należy stosować zarówno leki biologiczne, jak i biopodobne, analizując ich wykorzystanie także na podstawie danych efektywności kosztowej każdego leku, których w obecnej chwili brakuje. Wskaźnik efektywności kosztowej kumuluje w sobie zarówno bezpieczeństwo stosowania leku, możliwe powikłania, koszty wstępne leku, a także długoterminową poprawę jakości życia pacjentów dzięki jego użyciu.

![W Europie zanieczyszczenia powietrza powodują 800 tys. zgonów rocznie [WIDEO]](https://images.pb.pl/filtered/84b29d13-0727-4f42-8b21-de9566273376/76b0a951-3dc2-5503-975c-34480424e917_xc_l.jpg)
![Szansa na poprawę jakości życia dla pacjentów z HS [WIDEO]](https://images.pb.pl/filtered/08d54b79-36f0-499d-9e5e-731b6fdc1725/5e01a470-0cd4-51a2-9c4e-afeee2fc55b0_xc_l.png)